Pharmacomicrobiomics: the missing piece in precision medicine
In this article by Kathleen Shah, immunologist and translational scientist, we explore how the gut microbiome influences your response to medications.
Two patients. Same cancer diagnosis. Same drug, same dose, same oncologist. One responds. One does not, and develops severe, debilitating diarrhoea that forces a dose reduction, buying the tumour time. Same protocol. Completely different outcomes.
This is not a rare edge case. It is one of the most routine frustrations in clinical medicine, and it plays out across virtually every drug class: antidepressants that work for some and do nothing for others, blood thinners that require wildly different doses to hit the same therapeutic window, chemotherapies that produce life-threatening toxicity in a minority of patients who, on paper, look identical to those who tolerate them well.
We have spent decades building a framework for explaining this variability. It is a good framework, but it is incomplete. And what has been missing turns out to be living inside us.
Beyond the genome
The dominant answer to the variability problem has been pharmacogenomics (PGx): the idea that inherited differences in drug-metabolising enzymes and targets explain why people respond differently. For specific drugs, it genuinely does. CYP2D6 polymorphisms determine whether codeine is metabolised into morphine at all. VKORC1 and CYP2C9 variants underpin the notoriously narrow dosing window for warfarin, among many others.
These are real, validated, clinically useful insights. But across many drug classes (SSRIs, antihypertensives, most chemotherapies), genetic variants explain only a minority of observed variability in response and toxicity. For many high-stakes drugs, genotype provides little or no actionable signal at all. The explanation for the remainder wasn't genetic.
In 1993, eighteen cancer patients in Japan died. Prescribed sorivudine, a new antiviral, alongside oral 5-fluorouracil (5-FU) prodrugs, the combination turned lethal. Intestinal flora metabolised sorivudine to (E)-5-(2-bromovinyl)uracil (BVU); once absorbed systemically, BVU acted as a suicide substrate for dihydropyrimidine dehydrogenase (DPD), the hepatic enzyme responsible for clearing 5-FU. With DPD irreversibly disabled, 5-FU accumulated to toxic concentrations. Bone-marrow suppression. Catastrophic diarrhoea. Death. [1]
The microbiome was not on the risk model. A single microbial metabolic step, gut-generated but systemically lethal, killed eighteen people. The conceptual lesson was noted, and largely sidelined for another decade.
The field's formal account picks up in 2013, when Haiser and colleagues mapped a drug-microbiome interaction from organism to enzyme to in vivo pharmacokinetics for the first time. [2] Eggerthella lenta, a gut Actinobacterium, carries a cardiac glycoside reductase operon that inactivates digoxin, with the extent of inactivation modulated by luminal arginine availability and therefore by dietary protein intake. Diet. Microbe. Enzyme. Drug level. Clinical outcome. A complete causal chain.
In 2019, Zimmermann and colleagues scaled this question up by testing 76 human gut bacterial strains against 271 commonly prescribed oral drugs [3]. They found that two-thirds of these drugs (176/271) were chemically modified by at least one bacterial strain, implying that for most medications a clinician might prescribe, gut microbes are likely altering the drug before it even reaches the bloodstream. In principle, this could affect effective dose, making the amount of drug that becomes systemically available dependent on an individual's microbiome composition. However, not every microbial transformation will translate into a clinically meaningful effect: some structural changes will profoundly alter a drug's efficacy or toxicity, while others may be functionally negligible, and distinguishing between these scenarios requires careful, drug-by-drug investigation. What Zimmermann's study could not fully resolve was how much this varied between individuals. The following year, Javdan and colleagues demonstrated meaningful inter-individual variability in microbial drug metabolism across common compounds, reinforcing that what any drug encounters in the gut differs not just between species but between people[4].
How the microbiome alters drug behaviour
The gut microbiome encodes an estimated 150 times more genes than the human genome, [5] giving it enormous metabolic breadth. When an oral drug passes through the distal small intestine and colon, it moves through an environment where hundreds of microbial species carry out chemical reactions continuously.
1. Biotransformation
Gut bacteria chemically modify drugs via reductases, hydrolases, deconjugating enzymes and acetyltransferases, activating prodrugs, inactivating parent compounds, or generating toxic intermediates.
Irinotecan chemotherapy: Bacterial β-glucuronidases deconjugate SN-38G, a detoxified metabolite secreted into bile, back to its active cytotoxic form in the gut lumen, producing dose-limiting diarrhoea proportional to a patient's β-glucuronidase activity. Selective, non-absorbable inhibitors reduce this toxicity in animal models while preserving systemic drug exposure, improving the therapeutic index of an existing drug by targeting a single bacterial enzyme. [6, 7]
Parkinson's disease: Parkinson's disease is treated with levodopa, a dopamine precursor designed to cross the blood-brain barrier. The problem is that gut bacteria can intercept it first. Enterococcus faecalis expresses an enzyme that converts levodopa to dopamine in the intestinal lumen and a second bacterium, Eggerthella lenta, metabolises that dopamine further, compounding the loss before the drug reaches circulation. Levodopa is co-administered with carbidopa to prevent exactly this kind of peripheral conversion, but carbidopa targets the human enzyme. The bacterial version is structurally distinct enough to escape it. In rodent models, a small-molecule inhibitor designed specifically against the bacterial enzyme increased systemic levodopa exposure and improved brain delivery without touching the human pharmacological target, suggesting the two can be separated therapeutically [8]. In patients with Parkinson's disease, higher faecal abundance of E. faecalis and the microbial tyrDC gene has been associated with lower peak plasma levodopa concentrations, consistent with the mechanism and with the clinical reality that the same dose behaves very differently between patients [9]
Kidney transplantation: Patients with high gut abundance of Faecalibacterium prausnitzii consistently require higher tacrolimus doses, because gut bacteria convert tacrolimus to a metabolite with approximately 15-fold lower immunosuppressant activity. [10, 11] Where under-immunosuppression risks rejection and over-immunosuppression risks infection and malignancy, the clinical stakes are immediate.
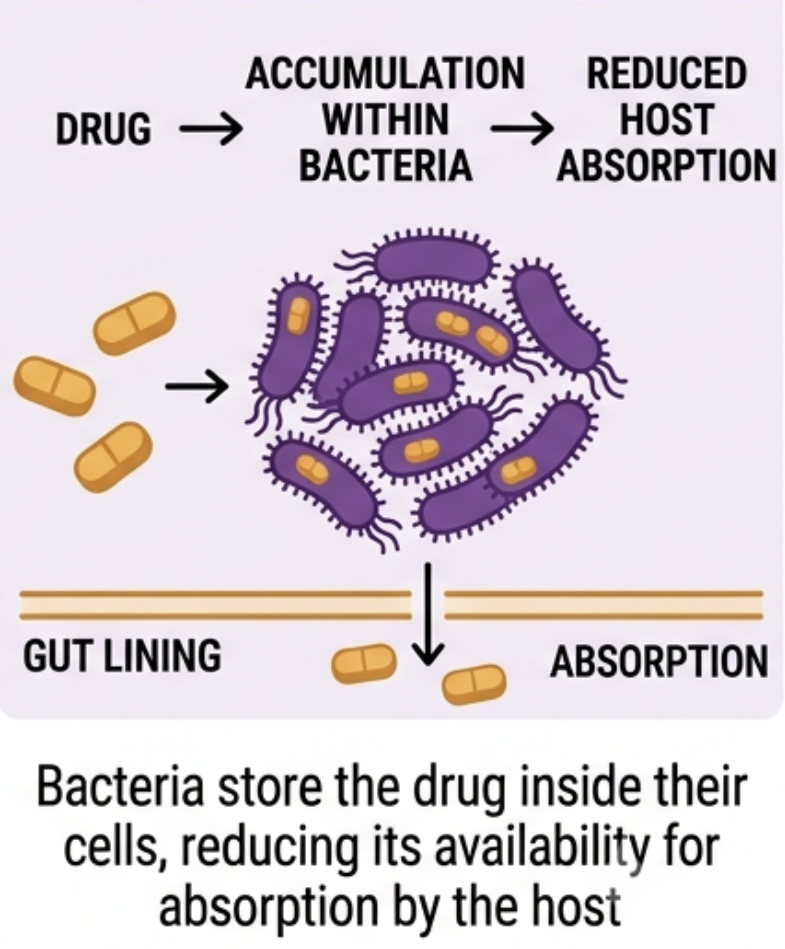
2. Bioaccumulation
Less appreciated, and more conceptually surprising, is a second direct mechanism: some bacteria sequester drugs intracellularly without chemically modifying them. The drug is not transformed; it disappears, removed from the intestinal lumen without leaving any metabolite that would register on standard pharmacokinetic screening.
Klünemann and colleagues identified 17 such interactions across 12 drugs and 25 bacterial strains, including duloxetine, one of the most widely prescribed antidepressants globally, alongside montelukast and rosiglitazone. [12] For duloxetine in particular, where clinical non-response is common and poorly understood, microbial bioaccumulation represents a plausible and currently unscreened contributor to variability. Unlike biotransformation, there is no metabolite to detect and no enzymatic target to inhibit. The drug simply fails to arrive.
3. Interference with host drug-metabolising enzymes
Every time a GP prescribes a broad-spectrum antibiotic to a patient on stable warfarin therapy, the INR may drift within days, not because the dose changed, but because antibiotics with high activity against Bacteroides fragilis reduce the gut's production of vitamin K2, shifting warfarin's anticoagulant balance. [13]
Behind this sits a broader system. Short-chain fatty acids (SCFAs) produced from dietary fibre fermentation regulate hepatic CYP450 expression; secondary bile acids activate FXR and PXR nuclear receptors governing xenobiotic-metabolising genes; tryptophan-derived indoles engage the aryl hydrocarbon receptor. Two patients with identical CYP genotypes can have meaningfully different functional enzyme activity depending on what their microbiome is producing.
Tryptophan metabolism governs a fork with direct implications for psychiatric pharmacology. Approximately 90% of the body's serotonin is produced in the gut, with synthesis directly regulated by microbial metabolites; when the microbiome preferentially routes tryptophan toward the kynurenine pathway instead, it generates neuroactive metabolites implicated in neuroinflammation and depression. [14] This is established biology. What is still emerging is the clinical translation: early data show that baseline microbiota profiles differ significantly between antidepressant responders and non-responders, and specific bacterial taxa have been proposed as pre-treatment targets for optimising response, though prospective evidence remains limited. [15, 16] For SSRIs, one of the most prescribed drug classes globally, the microbiome may shape both the biochemical substrate their mechanism depends on, and, as noted under bioaccumulation above, whether the drug arrives at all.
Metformin illustrates the bidirectionality. The drug consistently shifts microbial composition towards SCFA producers, alters bile acid metabolism and increases GLP-1 secretion, effects that contribute to its glycaemic benefit. Those same shifts are implicated in its diarrhoea and bloating. Efficacy and tolerability are two sides of the same microbial coin. Future microbiome profiling may identify which patients are likely to benefit from metformin through gut-mediated pathways and which will experience predominantly GI side effects, informing whether to start lower, titrate more slowly, or prioritise dietary co-intervention from the outset.
GLP-1 receptor agonists (semaglutide, liraglutide, tirzepatide) interact with a gut ecosystem that already shapes endogenous GLP-1 secretion through the same SCFA and bile acid pathways. [17, 18] Emerging data suggest the microbiome influences both the efficacy and GI tolerability of these drugs, with specific microbial signatures including Akkermansia muciniphila and SCFA producers associated with enhanced therapeutic response. [19]
4. Shaping the immune environment
Many modern drugs, particularly immunotherapies, depend on the immune system being in a specific state. The microbiome helps set that baseline through metabolites that modulate regulatory and effector T cells, innate lymphoid cells and myeloid activation.
Routy et al. and Gopalakrishnan et al., publishing simultaneously in Science in 2018, showed that baseline gut microbial diversity predicts anti-PD-1 response in melanoma and other epithelial cancers, with responder microbiomes enriched for Faecalibacterium and Akkermansia muciniphila. [20, 21] FMT (Faecal Microbiota Transplantation) from responding patients into germ-free mice restored sensitivity to checkpoint blockade. Antibiotic use during immunotherapy was associated with significantly worse outcomes. Early-phase trials show that FMT from responders can re-sensitise treatment-refractory melanoma patients, a causal relationship that has now moved into clinical protocols. Establishing the direction of effect is methodologically challenging, since disease states themselves reshape the microbiome; this is precisely why FMT experiments in germ-free mice have been so valuable: they establish causality, not just correlation. For clinicians, the practical question is shifting from whether the microbiome matters to which microbial profiles predict benefit, and whether those profiles can be modified before treatment begins.
What changes if we take this seriously
Unlike a genetic SNP (Single Nucleotide Polymorphism), the microbiome is not fixed. It shifts with diet, antibiotics, illness and time, and it can be deliberately altered. This transforms pharmacomicrobiomics from a predictive field into an interventional one: we can, in principle, modify a patient's microbial community to change how a drug behaves, shifting a non-responder toward response or reducing the microbial activity generating a toxic metabolite before prescribing begins.
What a patient eats, how antibiotic courses are managed around chronic medication, what their gut looks like before treatment starts: these become clinical variables rather than background noise.
Microbiome profiling alongside standard pre-prescribing assessment is the logical extension of existing clinical practice. Just as clinicians routinely check renal function or liver enzymes before initiating a new medicine, microbiome profiling may become a standard step for drugs where microbial interactions are clinically significant. [22] Current metagenomic platforms can resolve bacterial species and functional enzyme capacity within five to seven days at a cost comparable to standard pharmacogenomic panels; clinical interpretation frameworks are still being standardised. Machine learning algorithms trained on bacterial enzyme activity profiles can now predict susceptibility to biotransformation and bioaccumulation from drug structure, beginning to make individual risk stratification feasible before the first dose is given. [23]
The clearest near-term opportunity is oncology, and it is already moving. Microbiome profiling alongside tumour genomics and PD-L1 staining is already in clinical protocols for checkpoint inhibitor therapy. Patients beginning immunotherapy at centres running microbiome trials can today have their gut community assessed as part of treatment stratification. FMT from immunotherapy responders into refractory patients is in early-phase trials and producing signals.
Standardisation across sequencing platforms, universal clinical reference frameworks and regulatory pathways for microbiome-based diagnostics remain to be resolved. Routine integration into everyday prescribing is a 10-15 year horizon. In oncology, that horizon is already within sight.
The polypharmacy problem
We cannot yet adjust every prescription based on your microbiome. But the polypharmacy problem illustrates what is at stake if we do not. The average patient over sixty-five in the UK takes five or more medications simultaneously, and drug interaction checkers assess chemical incompatibilities between drugs, not microbial ones. Yet the gut microbiome is the shared metabolic infrastructure through which all of those drugs pass. A patient simultaneously on warfarin, metformin, a PPI, a statin and an antidepressant is creating a complex pharmacomicrobiomic environment: the PPI altering microbial diversity, the metformin shifting SCFA producers, bacterial enzyme activity modulating the effective exposure of every other drug on the list. Current prescribing practice cannot see these interactions. A framework that can would represent a fundamental advance in the safety of complex medication regimens.
Those two patients from the opening, same cancer, same drug, same oncologist, completely different outcomes, cannot yet be separated by any pharmacomicrobiomic test in routine clinical use. But the biology that explains the difference is no longer speculative. It sits in the mechanisms described above: in the bacteria metabolising the drug before it reaches its target, in the microbial community calibrating the immune environment the drug needs to function, in the gut ecosystem both patients brought to the clinic without anyone measuring it. The gut, it turns out, is one of the most pharmacologically active organs in the body. Learning to read it, and to work with it rather than around it, may be one of the defining clinical advances of the next generation.
For clinical practice: what to consider today
The full integration of pharmacomicrobiomics into prescribing is years away. These implications are actionable now.
- Unexplained INR drift in patients on stable warfarin should prompt review of recent antibiotic prescriptions and assessment of other potential confounders; the association between broad-spectrum antibiotic use and warfarin instability is consistent across cohorts, even where the precise mechanism remains under investigation.
- Erratic response to a previously stable drug regimen, particularly following antibiotics, significant GI illness, or major dietary change, may reflect microbiome disruption rather than disease progression or non-adherence.
- Long-term PPI co-prescription in patients on multiple gut-processed drugs warrants particular thought; the documented dysbiotic effects are not clinically neutral in a complex regimen.
- Every broad-spectrum antibiotic prescription is an uncontrolled pharmacomicrobiomic intervention, with downstream consequences for every other drug the patient takes.